
European Journal of Neurodegenerative Diseases 2024; 13(1) January-April: 29-31
SIMILARITIES IN THE PATHOGENESIS OF NEURODEGENERATIVE DISEASES AND THE DIFFICULTY WITH DIAGNOSIS
Letter to the Editor
E. Toniato *
Department of Innovative Technologies in Medicine and Dentistry, G. D’Annunzio University, Chieti, Italy.
*Correspondence to:
Prof. Elena Toniato,
Department of Innovative Technologies in Medicine and Dentistry,
G. D’Annunzio University,
Chieti, Italy.
e-mail: elena.toniato@unich.it
| Received: 6 January 2024 Accepted: 2 February 2024 |
2974-6345 (2024) Copyright © by BIOLIFE This publication and/or article is for individual use only and may not be further reproduced without written permission from the copyright holder. Unauthorized reproduction may result in financial and other penalties. Disclosure: all authors report no conflicts of interest relevant to this article. |
KEYWORDS: neurodegenerative disease, pathogenesis, diagnosis, protein, Alzheimer’s disease
INTRODUCTION
It is possible that diseases involving neuronal pathology share a similar pathogenesis, including inflammation and the aggregation of abnormal proteins. Molecular advances have shown that malformed proteins, such as amyloid beta (Aβ), cause damage to the brain by creating toxicity and inflammation (1,2). The deposition and aggregation of abnormal proteins in the brain is involved in some neurodegenerative diseases, including Alzheimer’s Disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD). Today the pathogenesis of these diseases is a little clearer, but it is not yet fully explained and there seems to be much more to discover. The diagnosis of neurodegenerative disease is often difficult to make, as diseases often share many similarities, and it can sometimes only be made after the patient’s death. The various pharmacological therapies available are limited and are often determined on the basis of the symptoms that the patient presents. However, the more we learn about these diseases, the more therapeutic tools can be constructed.
DISCUSSION
Neurodegenerative disorders are diseases that present central nervous system (CNS) dysfunction with the loss of neurons. The most common of these diseases are AD and PD, and the pathogenesis is mediated by protein dysfunction in both disorders. AD and PD present a similar pathogenic mechanism which concerns the alteration of proteins, their deposition, and their folding (3).
In AD, the protein dysfunction involves the misfolding of Aβ and the Tau protein, which leads to the formation of destructive Aβ plaques in the brain. In PD, there is misfolding of the protein alpha-synuclein (α-syn), which aggregates to form neuronal inclusions called Lewy bodies. This pathogenic mechanism is also present in Huntington’s disease (HD), where the huntingtin protein is misfolded and aggregated. This shows that many neurological disorders may present similar characteristics and therefore require common therapy.
Often, the symptoms and therapy of the different brain disorders overlap, making the diagnosis difficult, even if considerable progress has been made today with biomarkers. Neurodegenerative diseases can be classified based on clinical symptoms, anatomical region, biochemical modification of neuronal or glial proteins, and abnormalities of extracellular proteins. Clinical signs may include cognitive decline or alterations in CNS functions and/or dementia. The involved tissues may concern the limbic system, neocortical areas, hippocampus, and cortex. In addition, there are protein abnormalities and the loss of neurons, processes that can be highlighted with immunohistochemistry, which can help in the diagnosis.
The diagnosis should take the accumulation of both intracellular and extracellular proteins into consideration. Different proteins can be involved in neurodegeneration, which can be seen in the brains of elderly people presenting alterations in brain structures. The distribution of abnormal proteins in the brain and their characterization can be of great help in the classification of these diseases.
Misfolded proteins generate the formation of Aβ oligomers in the CNS and therefore, amyloidosis (Fig.1). Aβ oligomers were found in in vitro experiments using cell cultures from mouse models mimicking AD and brain tissue samples from post-mortem AD patients, demonstrating that these peptides are key components of amyloidosis and illness (4). It is hypothesized that in addition to amyloidosis, other abnormal protein molecules may also participate in brain disorders. Furthermore, as there are different types of Aβ formations, these can interact with each other, damaging neurons and causing disease.
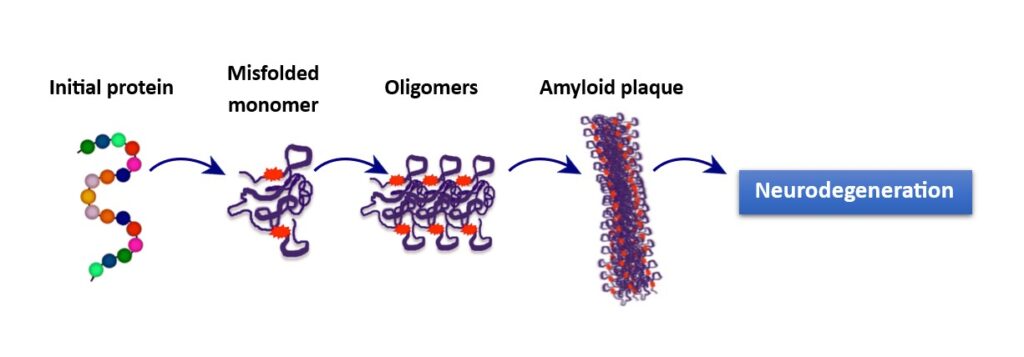
Fig. 1. Misfolded proteins are generated by an initial protein that forms a monomer. Monomers join to create oligomers which aggregate and form amyloid (Aβ) plaques, which damages neurons and leads to neurodegeneration.
Aggregated plaques of Aβ protofibrils cause neurotoxicity with neuronal damage and play an important role in the pathogenesis of neurodegenerative diseases such as AD. The accumulation of Aβ in the brain precedes the disease, which can manifest even after several years. The in-depth study of Aβ facilitates therapeutic research for these neurodegenerative diseases as there is much needed for resolving this dilemma that affects a substantial amount of people around the world.
CONCLUSIONS
Recently it has been observed that the inhibition of Aβ with proteases may be the right path for therapeutic research. For example, different studies utilizing the transport protein transthyretin (TTR), which binds and alters the augmentation of Aβ clustering, have shown that it inhibits the Aβ toxicity both in vitro and in vivo (5-7). However, more studies are needed to clarify many obscure points presented by these neurodegenerative diseases.
Conflict of interest
The author declares that they have no conflict of interest.
REFERENCES
- Varnum MM, Ikezu T. The Classification of Microglial Activation Phenotypes on Neurodegeneration and Regeneration in Alzheimer’s Disease Brain. Archivum Immunologiae et Therapiae Experimentalis. 2012;60(4):251-266. doi:https://doi.org/10.1007/s00005-012-0181-2
- Carrillo-Mora P, Luna R, Colín-Barenque L. Amyloid Beta: Multiple Mechanisms of Toxicity and Only Some Protective Effects? Oxidative Medicine and Cellular Longevity. 2014;2014:1-15. doi:https://doi.org/10.1155/2014/795375
- Forloni G, Terreni L, Bertani I, et al. Protein misfolding in Alzheimer’s and Parkinson’s disease: genetics and molecular mechanisms. Neurobiology of Aging. 2002;23(5):957-976. doi:https://doi.org/10.1016/S0197-4580(02)00076-3
- Kass B, Schemmert S, Zafiu C, et al. Aβ oligomer concentration in mouse and human brain and its drug-induced reduction ex vivo. Cell reports medicine. 2022;3(5):100630-100630. doi:https://doi.org/10.1016/j.xcrm.2022.100630
- Costa R, Gonçalves A, Saraiva MJ, Cardoso I. Transthyretin binding to A-Beta peptide – Impact on A-Beta fibrillogenesis and toxicity. FEBS Letters. 2008;582(6):936-942. doi:https://doi.org/10.1016/j.febslet.2008.02.034
- Stein TD, Anders NJ, DeCarli C, Chan SL, Mattson MP, Johnson JA. Neutralization of Transthyretin Reverses the Neuroprotective Effects of Secreted Amyloid Precursor Protein (APP) in APPSw Mice Resulting in Tau Phosphorylation and Loss of Hippocampal Neurons: Support for the Amyloid Hypothesis. Journal of Neuroscience. 2004;24(35):7707-7717. doi:https://doi.org/10.1523/jneurosci.2211-04.2004
- Du J, Cho PY, Yang DT, Murphy RM. Identification of beta-amyloid-binding sites on transthyretin. Protein Engineering Design and Selection. 2012;25(7):337-345. doi:https://doi.org/10.1093/protein/gzs026