
European Journal of Neurodegenerative Diseases 2017; 6(2) July-December: 12-16
NF-κB ACTIVITY IN ALZHEIMER’S DISEASE
Robuffo 1* and A. Palmieri 2
1 Institute of Molecular Genetics, National Research Council, Section of Chieti, Chieti, Italy;
2 Department of Experimental, Diagnostic and Specialty Medicine, University of Bologna, Bologna, Italy.
*Correspondence to:
Dr. Iole Robuffo,
Institute of Molecular Genetics,
National Research Council,
Section of Chieti,
Chieti, Italy.
e-mail: iole.robuffo@cnr.it
| Received: 02 September, 2017 Accepted: 07 October, 2017 |
2279-5855 (2017) Copyright © by BIOLIFE This publication and/or article is for individual use only and may not be further reproduced without written permission from the copyright holder. Unauthorized reproduction may result in financial and other penalties. Disclosure: all authors report no conflicts of interest relevant to this article. |
ABSTRACT
Alzheimer’s disease (AD) is a progressive neurodegenerative disease and is the most common type of dementia. In the USA, the disease affects approximately 5 million individuals, and it is expected that this number will increase drastically with the increasing age of the elderly population. Affected patients first experience mild memory loss which worsens as the disease progresses, and results in cognitive decline and ultimately, death. AD affects the brain with deposits of abnormal proteins and neuronal death. As of date, therapeutic options are limited and effective treatments still need to be discovered. Nuclear factor-κB (NF-κB) is a transcription molecule containing binding sites for genes involved in neuroinflammation and regulates genes that control cell replication and death. Gene therapy utilizing the signaling pathway of NF-kB in the brain could be targeted for gene therapy to treat AD. Blocking NF-kB could yield anti-inflammatory effects for the disease, leading to the suppression of growth hormone (GH), insulin-like growth factor (IGF-1), cell death, and also inflammatory cytokines such as IL-1, IL-6, and tumor necrosis factor (TNF) that are involved in the pathogenesis of AD.
KEYWORDS: Alzheimer’s disease, neurodegenerative disease, dementia, gene therapy, NF-κB, inflammation
INTRODUCTION
Alzheimer’s disease (AD) is a progressive brain disorder and is the most common type of dementia (1). At present, in the USA, approximately 5 million individuals are diagnosed with AD, and this figure could reach 13.8 million over the next 40 years, with the increasing age of the elderly population (2). Such an increase will come with a heavy clinical burden for society.
In this disease which can appear after the age of 65, there may be symptoms such as disorientation, mood swings, memory loss, problems with speech, depression, and hypertension (3). Furthermore, patients may present complications such as dehydration and infectious bacterial diseases (4). AD occurs particularly in elderly subjects with evidence of cerebral Lewy bodies and vascular alterations. Diagnosis can be made by utilizing cerebrospinal fluid (CSF) biomarkers and positron emission tomography (PET) (5). In recent years, the understanding of AD has seen considerable progress but there is still much to discover in this debilitating disease.
DISCUSSION
AD presents with amyloid plaques and neurofibrillary tangles (NFTs) in the central nervous system (CNS), with a decline in cognitive functioning and neuronal loss (6). The disease is caused by the improper cleavage of the amyloid precursor protein which forms abnormal β-amyloid (Aβ) which, by joining fibrils, forms Aβ plaques (7). The accumulation of abnormal proteins and the formation of plaques can last for several years before there are any symptoms of the disease.
In this disorder, amyloid angiopathies, neuritis, glial polyps, and glial response may be noted. These pathological signs cause neuronal and synaptic dysfunction with neurodegeneration and tissue atrophy. Aβ plaques are mainly formed by abnormally folded Aβ that are made up of 40 to 42 amino acids (8). The deposition of Aβ occurs in the iso-cortex, which influences the subcortex (9). The most abundant Aβ in the plaques is Aβ42, which is present in higher quantities than Aβ40 as it is less soluble (10). In the CNS, NFTs are formed by conjoined helical filaments made of hyperphosphorylated tau protein which is a microtubule-associated protein that stabilizes microtubules (11). This protein initiates and spreads in the medial temporal lobe cortex and hippocampus without invading motor, sensory, and visual areas. The severity of AD depends on the severity of neuronal and synaptic damage, and also the amount of Aβ formed (12). The genetic mutations that occur in AD are related to the formation of Aβ, leading to the generation of abnormal and pathological Aβ and resulting in neuronal dysfunction and neurodegeneration mediated by inflammation (13).
In this disease, neuroinflammation is involved and is decisive in the progression of neuropathological mutations (14,15). Activated immune cells are found within close proximity to, as well as inside, the plaques (16), and anti-inflammatory treatments have been seen to delay the onset of AD (17). Therefore, nonsteroidal anti-inflammatory drugs (NSAIDs) may help prolong the period before onset. NSAIDs block COX-2, resulting in inhibition of inflammatory prostaglandins that are induced by IL-1 and are involved in fever (18).
Nuclear factor kappa B (NF-kB) is a protein complex that functions as a transcription factor for protein production, including cytokines and chemokines. It is hypothesized that NF-kB is a key molecule that controls inflammation and apoptosis in many pathologies, including AD (19). Inflammation is known to play a key role in the pathophysiology of psychiatric diseases by involving pro-inflammatory cytokines such as IL-1 and tumor necrosis factor (TNF) (20,21). In AD, the plaques show signs of activated immunity and inflammation driven by IL-1 which, by binding to its receptor IL-1R on the cell membrane, leads to the formation of MyD88 with consequent activation of NF-kB and protein transcription (22) (Fig.1). TNF is also a highly inflammatory protein, which by binding to its receptor TNFR, activates IKKa, b, g, with consequent activation of NF-kB and transcription of the protein (23) (Fig.2).
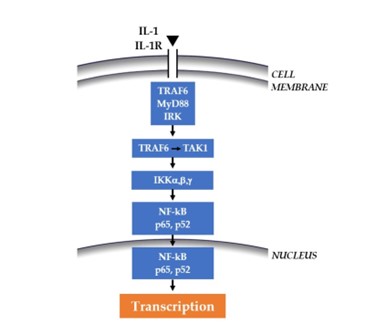
Fig. 1. By binding its receptor IL1-R, IL-1 activates the cascade that leads to NF-kB and the transcription of the protein.

Fig. 2. By binding its receptor TNFR, TNF activates the cascade that leads to NF-kB and the transcription of the protein.
Although there has been progress in the study of AD in recent years, classic therapeutic strategies have not been found to be effective. Therefore, much molecular research dedicated to the treatment of AD is concentrated on gene therapy, the insertion of foreign genes into cells to genetically modify diseased cells. NF-κB is a transcription molecule containing binding sites for genes involved in brain inflammation and regulates genes that control cell replication and death (24). Therefore, some inflammatory molecules that act through the signaling pathway of NF-kB at the brain level could be targeted. A possible anti-inflammatory therapeutic strategy in AD could involve the blocking of NF-kB, since its inhibition leads to the suppression of growth hormone (GH), insulin-like growth factor (IGF-1), and cell death (25). Furthermore, blocking NF-kB also suppresses inflammatory cytokines such as IL-1 (26), IL-6 (27), and TNF (28), which play an important role in the pathogenesis of AD (29).
CONCLUSIONS
Inflammation plays a key role in the progression of AD. Pro-inflammatory cytokines such as IL-1 and TNF that are found in plaques mediate neuroinflammation through the activation of NF-kB, aggravating the pathological state of the disease. The inhibition of these pro-inflammatory cytokines and other molecules, such as prostaglandins, would certainly help improve symptoms in AD.
Conflict of interest
The authors declare that they have no conflict of interest.
REFERENCES
- Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. The Lancet. 2011;377(9770):1019-1031. doi:https://doi.org/10.1016/s0140-6736(10)61349-9
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia. 2016;12(4):459-509. doi:https://doi.org/10.1016/j.jalz.2016.03.001
- Tarawneh R, Holtzman DM. The Clinical Problem of Symptomatic Alzheimer Disease and Mild Cognitive Impairment. Cold Spring Harbor Perspectives in Medicine. 2012;2(5):a006148-a006148. doi:https://doi.org/10.1101/cshperspect.a006148
- Maheshwari P, Eslick GD. Bacterial Infection and Alzheimer’s Disease: A Meta-Analysis. Journal of Alzheimer’s Disease. 2014;43(3):957-966. doi:https://doi.org/10.3233/jad-140621
- Landau SM, Lu M, Joshi AD, et al. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of β-amyloid. Annals of Neurology. 2013;74(6):826-836. doi:https://doi.org/10.1002/ana.23908
- Nelson PT, Braak H, Markesbery WR. Neuropathology and Cognitive Impairment in Alzheimer Disease: A Complex but Coherent Relationship. Journal of neuropathology and experimental neurology. 2009;68(1):1-14. doi:https://doi.org/10.1097/NEN.0b013e3181919a48
- O’Brien RJ, Wong PC. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annual Review of Neuroscience. 2011;34(1):185-204. doi:https://doi.org/10.1146/annurev-neuro-061010-113613
- van der Kant R, Goldstein LS. Cellular Functions of the Amyloid Precursor Protein from Development to Dementia. Developmental Cell. 2015;32(4):502-515. doi:https://doi.org/10.1016/j.devcel.2015.01.022
- Kimura T, Yamashita S, Fukuda T, et al. Hyperphosphorylated tau in parahippocampal cortex impairs place learning in aged mice expressing wild-type human tau. The EMBO Journal. 2007;26(24):5143-5152. doi:https://doi.org/10.1038/sj.emboj.7601917
- Gu L, Guo Z. Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. Journal of Neurochemistry. 2013;126(3):305-311. doi:https://doi.org/10.1111/jnc.12202
- Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Molecular Neurodegeneration. 2009;4(1):13. doi:https://doi.org/10.1186/1750-1326-4-13
- Sadigh-Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J. Amyloid-beta: a crucial factor in Alzheimer’s disease. Medical principles and practice : international journal of the Kuwait University, Health Science Centre. 2015;24(1):1-10. doi:https://doi.org/10.1159/000369101
- Giri M, Zhang M, Lü Y. Genes associated with Alzheimer’s disease: an overview and current status. Clinical Interventions in Aging. 2016;11:665. doi:https://doi.org/10.2147/cia.s105769
- Morales I, Guzmán-Martínez L, Cerda-Troncoso C, Farías GA, Maccioni RB. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Frontiers in Cellular Neuroscience. 2014;8(112). doi:https://doi.org/10.3389/fncel.2014.00112
- Zilka N, Ferencik M, Hulin I. Neuroinflammation in Alzheimer’s disease: protector or promoter?. Bratislavské lekárske listy. 2006;107(9-10):374-383.
- Guillot-Sestier MV, Town T. Innate Immunity in Alzheimer’s Disease: A Complex Affair. CNS & Neurological Disorders – Drug Targets. 2013;12(5):593-607. doi:https://doi.org/10.2174/1871527311312050008
- Breitner JC, Welsh KA, Helms MJ, et al. Delayed onset of Alzheimer’s disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiology of Aging. 1995;16(4):523-530. doi:https://doi.org/10.1016/0197-4580(95)00049-k
- Bacchi S, Palumbo P, Sponta A, Coppolino MF. Clinical Pharmacology of Non-Steroidal Anti-Inflammatory Drugs: A Review. Anti-Inflammatory & Anti-Allergy Agents in Medicinal Chemistry. 2012;11(1):52-64. doi:https://doi.org/10.2174/187152312803476255
- Baker RG, Hayden MS, Ghosh S. NF-κB, Inflammation, and Metabolic Disease. Cell Metabolism. 2011;13(1):11-22. doi:https://doi.org/10.1016/j.cmet.2010.12.008
- Miller AH, Maletic V, Raison CL. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biological Psychiatry. 2009;65(9):732-741. doi:https://doi.org/10.1016/j.biopsych.2008.11.029
- Mittleman BB, Castellanos FX, Jacobsen LK, Rapoport JL, Swedo SE, Shearer GM. Cerebrospinal fluid cytokines in pediatric neuropsychiatric disease. Journal of Immunology. 1997;159(6):2994-2999. doi:https://doi.org/10.4049/jimmunol.159.6.2994
- Shaftel SS, Griffin WST, O’Banion MK. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. Journal of Neuroinflammation. 2008;5(1):7. doi:https://doi.org/10.1186/1742-2094-5-7
- Wajant H, Scheurich P. TNFR1-induced activation of the classical NF-κB pathway. FEBS Journal. 2011;278(6):862-876. doi:https://doi.org/10.1111/j.1742-4658.2011.08015.x
- Baldwin AS. Series Introduction: The transcription factor NF-κB and human disease. The Journal of Clinical Investigation. 2001;107(1):3-6. doi:https://doi.org/10.1172/JCI11891
- Tilstra JS, Clauson CL, Niedernhofer LJ, Robbins PD. NF-κB in Aging and Disease. Aging and disease. 2011;2(6):449-465.
- Griffin WS, Stanley LC, Ling C, et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(19):7611-7615. doi:https://doi.org/10.1073/pnas.86.19.7611
- Hüll M, Berger M, Volk B, Bauer J. Occurrence of Interleukin‐6 in Cortical Plaques of Alzheimer’s Disease Patients May Precede Transformation of Diffuse into Neuritic Plaquesa. Annals of the New York Academy of Sciences. 1996;777(1):205-212. doi:https://doi.org/10.1111/j.1749-6632.1996.tb34420.x
- van der Wal EA, Gómez-Pinilla F, Cotman CW. Transforming growth factor-β1 is in plaques in Alzheimer and Down pathologies. NeuroReport. 1993;4(1):69-72. doi:https://doi.org/10.1097/00001756-199301000-00018
- Tak PP, Firestein GS. NF-κB: a key role in inflammatory diseases. Journal of Clinical Investigation. 2001;107(1):7-11. doi:https://doi.org/10.1172/jci11830